

Oleh: Susan E. Detmer
Diterjemahkan oleh: Agna D. Lantria
DISCLAIMER
This article was translated to enable Bahasa Indonesia readers to better understand the topic explained inside the article in their native language. The translator made no profit in translating the article.
Pengenalan
Virus influenza A (AIV) adalah penebab utama penyakit respiratori tahunan pada babi di semua kelompok umur. Pada kasus yang tidak kompleks, kasusnya bersifat ringan dan self-limiting (sembuh sendiri) [67]. Serupa dengan penyakit pada manusia, influenza terjadi sepanang tahun dengan puncak-puncak yag terjadi musiman saat temeratur lingkungan dan kelembaban sedang [70].
Sejarah
Meskipun gejala klinis influenza pertama kali dikenali pada babi selama terjadinya pandemi flu spanyol tahun 1918 [36], isolasi pertama virus AI dari babi tidak terjadi sampai tahun 1930 [73]. virus H1N1 Influenza/A/swine/Iowa/15/1930 dianggap sebagai prototipe virus H1N1 klasik (cH1N1) yang secara bertahap berevolusi di Amerika Utara selama abad ke-20, sampai masuknya kaset gen internal tripel reasortansi (TRIG) yang terdiri dari gen virus AI yang berasal dari unggas, manusia dan babi [91]. Sejak masuknya kaset gen TRIG, telah terjadi ledakan keragaman genetik virus AI pada babi di seluruh dunia, dengan adanya perkembangan beberapa klaster genetik dan antigenik berbeda (untuk informasi lebih lanjut tentang evolusi virus AI pada babi lihat Bab 18).
Virologi
Influenza endemik pada babi pada umumnya disebabkan oleh virus AI dari subtipe H1N1, H1N2 dan H3N2. Infeksi yang disebabkan virus AI dengan asal seluruhnya dari manusia dan asal unggas dan reasortan berakibat munculnya subtipe tidak biasa pada babi telah ditemukan [29, 48, 49, 60]. Akan tetapi, hal ini terjadi secara sporadis dan tampaknya tidak mungkin berakibat munculnya infeksi endemik yang berkelanjutan. Serupa dengan itu, antibodi terhadap virus influenza B telah terdeteksi pada babi di Inggris dan China, tetapi virus influenza B belum pernah diisolasi dari babi [5, 54]. Meskipun jarang terjadi, virus influenza C (ICV) telah dideteksi pada babi [5, 54, 89]. Virus baru yang mirip dengan virus influenza C pada manusia sampai saat ini masih jarang ditemui pada babi akan tetapi mungkin memiliki sirkulasi yang lebih luas pada sapi [24, 71].
Pada level seluler, infeksi virus AI diinisiasi dengan berikatannya protein hemaglutinin (HA) pada permukaan virus dengan gula asam sialik pada permukaan sel epitel saluran pernafasan [26]. Ketika sudah masuk ke dalam sel, virus kemudian bereplikasi dan banyak partikel virus kemudian dilepaskan. Lesi mikroskopis mengindikasikan bahwa proses ini dapat terlihat dalam waktu 24 – 48 setelah infeksi. Tingkat progresi dan keparahan penyakit respirasi bervariasi tergantung virus AI nya, dan juga dapat dimodifikasi berdasarkan sejumlah faktor yang berbeda.
Mekanisme infeksi yang sebenarnya terjadi tetap konsisten, dan variabilitas dapat terlihat pada isolasi virus dari sampel hewan. Sel Madin-Darby canine kidney (MDCK) memiliki sensitivitas tertinggi untuk menumbuhkan virus AI asal babi, dan paling sering digunakan dalam penelitian dan aplikasi diagnostik [55]. Sel testis babi dan embrio ayam juga direkomendasikan selain sel MDCK. Kebanyakan virus AI dapat dipropagasi dalam garis sel ini dengan kondisi kultur standar, tetapi terdapat variabel antar strain. Beberapa virus AI tumbuh dalam waktu 48 hari, akan tetapi beberapa lainnya memerlukan waktu 5 – 7 hari atau pasase kedua untuk dapat diisolasi. Jumlah pasase untuk sel MDCK juga dapat memberikan pengaruh. Beberapa virus AI tumbuh sampai titer yang tinggi dalam sel MDCK hanya dengan jumlah pasase rendah (dibawah 100 kali pasase), sementara virus AI lainnya bertumbuh dengan titer yang tinggi dalam sel MDCK dengan jumlah pasase yang lebih tinggi (lebih dari 200 kali pasase). variasi-variasi yang teramati secara in-vitro ini tidak teramati secara in-vivo, dan mewakili salah satu inkongruensi antara hasil eksperimen in-vitro dan in-vivo.
Patogenisitas
Patogenisitas virus AI pada babi bisa jadi cukup bervariasi dan seringkali saat virus AI diisolasi saat terjadinya epizootik dalam peternakan, laporan anedoktal dari lapangan menunjukkan kematian yang lebih tinggi daripada ketika virus diuji inokulasi secara eksperimental dibawah kondisi yang dikendalikan [50]. Kemampuan virus AI untuk menyebabkan penyakit pada babi ditentukan dengan kombinasi faktor hospes, virus dan lingkungan. Faktor lingkungan dan permasalahan dalam manajemen pemeliharaan akan didiskusikan dengan lebih mendetail dalam bagian yang membahas tentang epidemiologi dalam bab ini. Dua dari faktor hospes yang harus diperhatikan adalah umur dan status imunitas babi. Imunitas dan vaksinasi akan didiskusikan dengan lebih detail dalam bab 17 dan 19.
Seperti kebanyakan spesies lainnya, anak babi baru lahir adalah salah satu populasi yang paling rentan terhadap virus AI, populasi ini mungkin memiliki atau tidak memiliki antibodi maternal (MDA) yang terdapat dalam kolostrum induk untuk melindungi mereka terhadap virus AI. Anak babi biasanya disapih pada umur 21-28 haru dan dipindahkan ke dalam kandang pemeliharaan dimana babi yang berasal dari induk yang berbeda dibesarkan bersama, dan kemudian dipindahkan lagi ke kandang grower-finisher ketika mencapai umur 10-12 minggu. Anak babi dalam sapihan sangat rentan terhadap sejumlah patogen pernafasan, dan rentan pula terhadap infeksi endemik dari sejumlah patogen yang dihubungkan dengan Porcine Respiratory Disease Complex (PDRC). PDRC adalah penyakit yang komplek dan multifaktorial yang didalamnya juga mencakup virus AI. Penyakit ini memiliki baik faktor infeksius maupun non-infeksius yang berkontribusi terhadap penyakitnya. Pada umumnya terlihat pada babi umur grower-finisher antara umur 3 sampai 6 bulan [63]. Seperti infeksi virus AI yang berdiri sendiri, PDRC dipengaruhi oleh kombinasi faktor hospes, patogen, dan lingkungan.
Satu faktor yang spesifik dari virus AI yang tergantung baik pada hospes dan virus adalah pengikatan HA dengan gula asam sialik. Spesifisitas hospes untuk virus AI ditentukan oleh tipe dan distribusi sel reseptor hospes dan struktur protein HA. Telah diterima secara umum bahwa virus AI manusia dan virus AI babi berikatan pada reseptor asam sialik NeuAcalpha2,6 Gal-linked (alpha2,6), dan virus AI avian berikatan dengan reseptor NeuAcalpha 2,3 (alpha2,3) [26]. Distribusi reseptor alpha 2,5 dan alpha 2,3 telah ditemukan baik pada manusia dan babi, alpha 2,6 tersebar di sepanjang saluran pernafasan dan alpha 2,3 lebih spesifik di alveoli [57, 77].
Teknik histokimia lektin (LH), yang digunakan untuk menentukan secara kualitatif lokasi reseptor asam sialik dalam saluran respirasi, telah menyebabkan perbedaan dalam distribusi reseptor influenza dan tipe sel spesifik yang terlibat, demikian juga perbedaan antar sel yang terinfeksi di dalam kultur dibandingkan dengan yang diprediksikan oleh LH dalam saluran pernapasan manusia [53, 61, 62, 72, 87] dan dalam saluran pernapasan babi [31, 76, 78]. Selain itu, cara masuk dan replikasinya virus AI yang telah dihilangkan asam sialik dari permukaannya masih belum diketahui [75].
Selain spesifisitas hospes, karakteristik virus yang virulen meliputi kemampuan untuk mempertahankan replikasi virus dalam level tinggi dan atau memperpanjang waktunya, mempertahankan replikasi dalam saluran pernafasan bawah, dan induksi ekspresi sitokin yang tinggi [32]. Sitokin yang diperhatikan meliputi interferon (IFN)-𝛼, faktor nekrosis tumor (TNF)-𝛼 dan interleukin (IL) 1, 6 dan 8. Induksi ekspresi berlebihan dari IL-1β, IL-8, dan TNF-𝛼 telah terlihat pada penyakit pernafasan yang ditingkatkan oleh vaksin (VAERD – vaccine-associated enhanced respiratory disease), yang ditandai dengan penyakit pernafasan berat disertai kesalahan vaksinasi dan selanjutnya diikuti infeksi virus [68].
Kapabilitas virulensi virus AI yang diturunkan berhubungan dengan interaksi protein virus baik dengan hospes maupun dengan satu sama lain. Protein virus yang paling banyak dipelajari adalah protein HA permukaan yang bertanggung jawab untuk perlekatan sel hospes, internalisasi dan fusi antara capsid virus dan membran endosom. Virus asal manusia dan babi tidak memiliki situs pembelahan multi basic yang ditemukan pada virus-virus AI ber patogenitas tinggi (subtipe HA H5 dan H7) yang telah dihubungkan dengan infeksi letal pada mencit dan musang, akan tetapi babi tampaknya memiliki kerentanan yang rendah [43]. Dua mutasi HA berbeda, E119G/V1521/N224K/Q226L dan N224K/Q226L, telah menunjukkan perubahan preferensi pengikatan dari 𝝰2,3 menjadi 𝝰2,6 dalam virus asal unggas [30], tetapi mutasi tambahan yaitu N158D atau N158K diperlukan untuk meningkatkan replikasi virus pada mutan, dan mutasi T318 tampaknya mampu menstabilisasi protein HA selama penelitian transmisi virus [28].
Tiga protein polymerase, PA, PB1 dan PB2 membentuk kompleks protein dan replikasi genom RNA-nya memainkan peranan kunci dalam tingkat replikasi virus dan genetic drift. Protein PB1-F2 yang merupakan hasil dari kerangka bukaan alternatif protein PB-1 dapat diidentifikasi pada beberapa virus AI asal babi [64]. Protein ini paling sering dihubungkan dengan induksi apoptosis sel sistem imun bawaan, tetapi juga dihubungkan dengan supresi respons interferon awal dalam sel yang terinfeksi, dan juga dalam peningkatan peradangan di jaringan [32].
Kebanyakan penelitian yang mempelajari tentang fungsi protein virus telah dilakukan dalam kultur sel dan model hewan laboratorium. Hanya sedikit penelitian yang telah berhasil mereplikasi hasil yang sesuai dengan asal hospes sesuai spesiesnya, yang merefleksikan relevansi hasil penelitian dan adanya batasan model hewan laboratorium untuk digunakan dalam mempelajari virus AI.
Sistem hospes laboratorium
Idealnya, virus AI harus dipelajari dalam hospes alaminya, dan babi domestik merupakan model laboratorium yang sangat baik baik untuk virus AI yang berasal dari babi maupun manusia [68]. Akan tetapi, model tikus untuk virus influenza masih sangat sering digunakan, meskipun terdapat fakta bahwa strain virus harus diadaptasikan terlebih dahulu pada mencit untuk dapat menyebabkan penyakit. Mencit yang dipingsankan dapat dipilih untuk mempelajari respons imun seluler, akan tetapi harus diperhatikan interpretasi hasil penelitian terhadap spesies yang lain karena mungkin hasilnya tidak bisa langsung diaplikasikan. Babi guinea merupakan model pengerat berbeda yang tidak mengharuskan adaptasi virus, tetapi virus lesi paru terbatas kecuali jika dosis virus tinggi [46]. Untuk virus AI asal babi yang diinfeksikan pada Babi guinea hanya terdapat transmisi terbatas, yang mungkin menjadi faktor yang membatasi penggunaan hewan model ini untuk mempelajari virus asal babi [46].
Model musang telah digunakan untuk mengukur patogenisitas virus AI dari berbagai spesies, termasuk babi. Model tersebut telah digunakan untuk model transmisi langsung dan tidak langsung, untuk mengamati potensi faktor virulensi dan respons imun, serta untuk membuat antibodi standar untuk uji inhibisi hemaglutinasi yang digunakan untuk manusia dan babi [90]. Dibandingkan dengan manusia dan babi, musang memiliki distribusi reseptor 𝝰2,3 sampai 𝝰2,6 dan bahkan pola lesi dan penyakit klinis yang serupa [77, 90]. Meskipun musang adalah model yang cocok, ia pun memiliki batasan yang terkait dengan interpretasi hasil eksperimental.
Jaringan pernafasan yang langsung diambil dari babi (explant) dapat dipertahankan dalam kamar inkubasi selama kurang lebih 48 jam dalam rangka mempelajari agen infeksius [17]. Meskipun reaksi sistemik penuh tidak dapat muncul dalam explant, beberapa virus dapat dipelajari dalam bagian-bagian jaringan terpisah dari satu hewan yang sama. Eksplan jaringan menawarkan transisi intermediat yang sangat baik dari kultur sel primer atau immortal menuju hewan model untuk penyakit. Model ini juga memberikan kesempatan untuk mengurangi jumlah hewan yang diperlukan untuk penelitian, yang dianjurkan oleh komite etika penelitian.
Penyakit klinis
Gejala klinis penyakit pernafasan akut yang disebabkan virus AI pada babi secara alami maupun yang diinokulasi virus secara eksperimental meliputi demam, anoreksia, batuk, susah bernafas dan”thumping” (suara keras yang dikeluarkan babi ketika seluruh badannya bergetar karena berusaha bernafas), bersin, leleran hidung, dan penambahan berat badan yang berkurang. Demam adalah gejala klinis paling konsisten, dan puncaknya dalam waktu 24 – 48 jam infeksi untuk sebagian besar strain virus AI. Gejala klinis mulai muncul paling cepat 1 hari setelah infeksi (DPI) seperti yang direkam dalam infeksi eksperimental pada babi dengan isolat virus AI baik yang diisolasi dari manusia maupun babi [4, 21, 22, 25-27, 34, 40, 41, 51, 66, 74, 79, 85], juga di hari ke-2 [3] dan ke-3 [33-35], bersamaan dengan waktu terdeteksinya virus dalam sekresi hidung.
Gejala klinis berhenti muncul di hari ke-4 dan 8 pada hewan yang diinfeksi secara eksperimental [4, 21, 22, 25-27, 34, 40, 41, 66, 74, 79, 85]. Pada hewan yang telah memiliki proteksi kuat dari vaksin sebelum dari vaksinasi sebelum diuji tantang dengan isolat virus yang mirip secara antigenik, jumlah virus yang didedahkan berkurang, dan pendedahan virus berakhir pada 2 -4 DPI [12, 26, 27, 79-81]. Vaksinasi homolog idealnya berakibat hanya sedikit virus yang didedahkan atau tidak ada virus sama sekali dan tidak ada gejala klinis, akan tetapi hal ini tidak dapat dijamin [1, 69].
Influenza pada babi adalah penyakit dengan morbiditas tinggi dan mortalitas rendah. Meskipun virus dapat bersirkulasi dalam peternakan tanpa keberadaan gejala klinis [9], akan tetapi angka kematian dapat teramati pada babi sebesar 80-100% pada saat wabah influenza dalam satu peternakan, dan sebesar 30-50% pada babi yang ada dalam peternakan yang telah terinfeksi secara endemik. Kematian karena infeksi influenza yang tidak mengalami komplikasi sangat jarang ditemukan pada babi, akan tetapi telah terlihat pada kasus diagnostik dimana batuk dapat menyebabkan hematoma trakea yang menyebabkan aspikisasi, dan dimana anoreksia menyebabkan pendarahan ulcer lambung dan kemudian eksanguinasi.
Seringkali angka kematian karena influenza pada babi karena infeksi bakterial sekunder atau ko-infeksi dengan PRDC. Ko-infeksi virus influenza bersama Haemophilus parasuis atau porcine reproductive and respiratory syndrome virus (PRRSV) dihubungkan dengan penyakit yang lebih berat pada babi [82, 83]. Patogen PDRC lain, Mycoplasma hyopneumoniae telah diperlihatkan mampu memiliki efek sementara atau tidak ada efek sama sekali terhadap hasil keseluruhan kon-infeksi dengan virus AI [88]. M. hyopneumoniae menyebabkan stasis silia dalam pohon trakea dan bronkhi, dan karena itu menghalangi mekanisme pertahanan melalui pengeluaran benda-benda asing dengan mukosilia. Virus AI adalah partikel berukuran kecil (80-120 nm) berarti bahwa virus tersebut dapat menghindari sel mukosilia, sehingga sinergi virus dengan patogen PRDC ini tidak mungkin terjadi.
Salah satu dari isolat paling virulen di lapangan dilaporkan menyebabkan angka mortalitas sebesar 10% pada babi finisher, akan tetapi PRRSV, Pasteurella multocida, Streptococcus suis, dan spesies Streptococcus lain juga terdeteksi pada paru [50]. Semua patogen ini dianggap bagian dari PRDC [63]. Karena virus AI adalah salah satu komponen pokok PRDC, gejala klinis PRDC serupa dengan gejala yang digambarkan disebabkan influenza. Kompleks penyakit ini dicirikan dengan morbiditas 30-70% dan mortalitas 4-6%, serupa dengan penyakit yang hanya disebabkan virus AI saja [63].
Patologi virus AI pada babi
Patologi Mikroskopis
Tanda-tanda lesi mikroskopis infeksi AI adalah bronkhitis nekopurulen dan bronkiolitis [13, 32]. Seperti ditunjukkan dalam gambar 16.1a, bronkioli normal memiliki lapisan sel epitelial tipis yang memiliki silia apikal dan sejumlah kecil jaringan limfoid peribronkiolar. Lesi flu awal terlihat paling cepat 24 jam pasca infeksi (PI) dan meliputi degenerasi vakuolar dan nekrosis sel epitel dengan menghilangnya silia apikal (Gambar 16.1b). Hal ini bersamaan dengan temuan ultrastruktural tunas-tunas virus yang banyak setelah 24 jam pasca infeksi [37]. 48 jam pasca infeksi, sel epitel nekrotik yang mengelupas mengumpul dalam lumen jalur udara bersama netrofil yang bermigrasi melewati epitelium, menciptakan lesi khas bronkiolitis nekropurulen. 72 jam pasca infeksi (gambar 16.1c), pengelupasan sel epitel nekrotik tampak lebih jelas, bersama campuran sel radang dalam jumlah kecil. Sel epitel yang tersisa menyebar menutupi membran basal (atenuasi), dan jaringan limfoid peribronkiolar meluas, dengan meningkatnya jumlah limfosit bercampur dengan beberapa makrofag (limfoid hiperplasia).
Antara hari ke-4 dan 5 pasca infeksi, tanda awal rekoveri meliputi beragam derajat hiperplasia epitelial, badan mitotik dalam sel epitel (Gambar 16.1d) dan keradangan ringan dalam bronki dan bronkioli seiring menyebarnya keradangan, memperlebar septa alveolar. Pada hari ke-7 sampai 10 pasca infeksi terdapat berbagai derajat penumonia interstitial, proliferasi limfoid perivaskular dan peribronkiolar, dan epitel bronkial dari normal sampai hiperplastik. Pada hari ke 14 – 21 pasca infeksi, jaringan respirasi yang rusak seharusnya sudah pulih kembali pada level mikroskopis [4, 18, 33, 35, 41, 56, 66, 74]. Selama proses pemulihan, bronkiolitis obliteran dapat merupakan akibat dari terpaparnya lamina propria dan formasi polip dalam lumen bronkiolar [32].
Pada beberapa kasus, replikasi virus dan kerusakan terbatas pada pohon bronkial, dan terdapat lobulus tanpa lesi, atau lesi terbatas pada bronkioli (Gambar 16.21 dan 16.2b). Pada kasus lainnya, peradangan meluas keluar bronkioli ke dalam dinding alveolar, menyebabkan lesi berat dengan konsolidasi lobular (Gambar 1.2c dan 16.rd). Meskipun lesi paling berat dan paling konsisten adalah pada bronkioli primer, lesi alveolar cukup bervariasi dan dapat meliputi ateletaksis sebelum tersumbatnya jalan udara, dan pneumonia interstitial, atau kombinasi dari lesi-lesi itu. Pola lesi alveolar lebih cenderung lobular. Hal ini karena kombinasi pola percabangan pohon bronkial yang mungkin berakibat distribusi virus secara ireguler, dan septa interlobular yang menebar ditemukan pada babi dan sapi mencegah persebarannya ke lobulus yang berdekatan. Kolaps alveolar atau atelektasis obstruktif tidak selalu diperhatikan baik di level mikroskopis maupun makroskopis, dan terjadi saat terdapat akumulasi debris yang cukup signifikan (atau bronkiolitis obliteran) yang menyumbat aliran udara ke dalam celah alveolar.
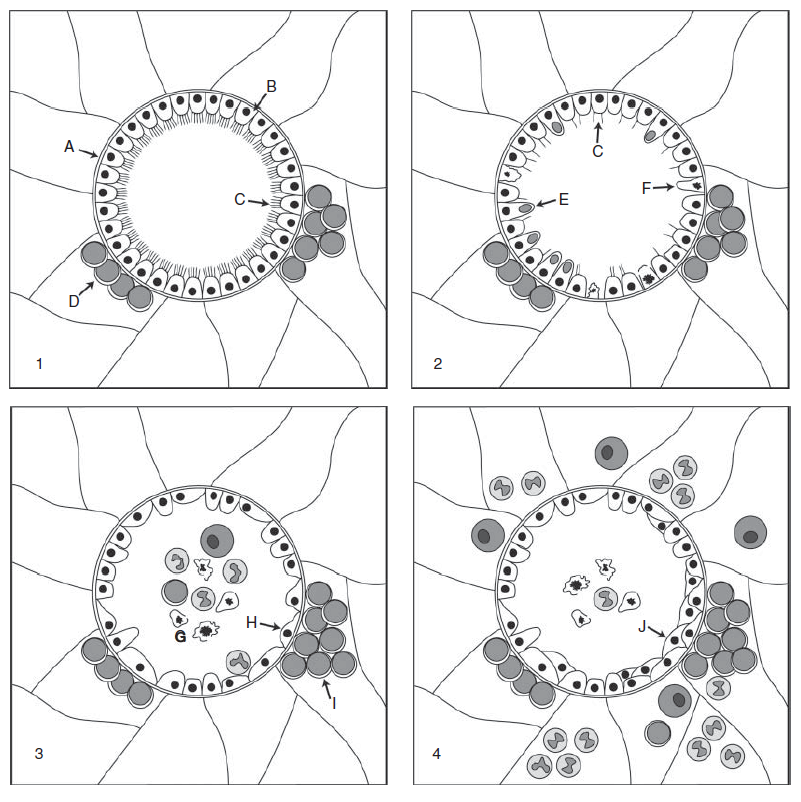
Gambar 16.1 Bronkioli pulmoner selama tahapan infeksi virus AI pada babi. Sumber Juliane Dubner dan University Saskatchewan. Gambar 16.1a. Bronkioli normal (A) yang memiliki lapisan sel epitelial tipis (B) dengan silia apikal ( c) dan jaringan limfoid peribronkiolar (D). Gambar 16.1b. Lesi awal influenza berupa degenerasi vakuolar (E) dan nekrosis (F) sel epitel dengan hilangnya silia apikal terlihat paling cepat 24 jam pasca infeksi (PI). Gambar 16.1c. Lesi influenza setelah 72 jam pasca infeksi dengan sel epitel yang mengelupas dan nekrosis, sejumlah sel radang (G), atenuasi sel epitel yang tersisa (H), dan hiperplasia limfoid ringan (I). Gambar 16.1d. Lesi influenza – 7 hari pasca infeksi, menunjukkan berbagai derajat hiperplasia sel epitel (J), dan figur mitotik dalam beberapa sel epitel.
Proses yang mendasari formasi pneumonia intersitital diidentifikasi menggunakan pemeriksaan ultrastruktural 5 jam setelah infeksi, yang menunjukkan virus AI yang muncul dari sel pneumosit tipe II [86]. Pengelupasan sel epitel alveolar bersama edema dan fibrin yang memisahkan sel interstitial teramati dalam 24 jam pasca inokulasi [86]. Selama 24 – 48 jam pasca inokulasi, terjadi kehancuran pneumosit dan migrasi netrofil diikuti oleh limfosit dan makrofag, dengan konsolidasi alveolar memanjang dari bronkioli ke bronkioli dalam lobulus. Pneumonia interstitial dicirikan oleh septa alveolar yang menebal karena pneumosit tipe II yang membengkak, penggantian sel pneumosit tipe II dan I yang mengelupas, edema interstitial dan akumulasi monosit.
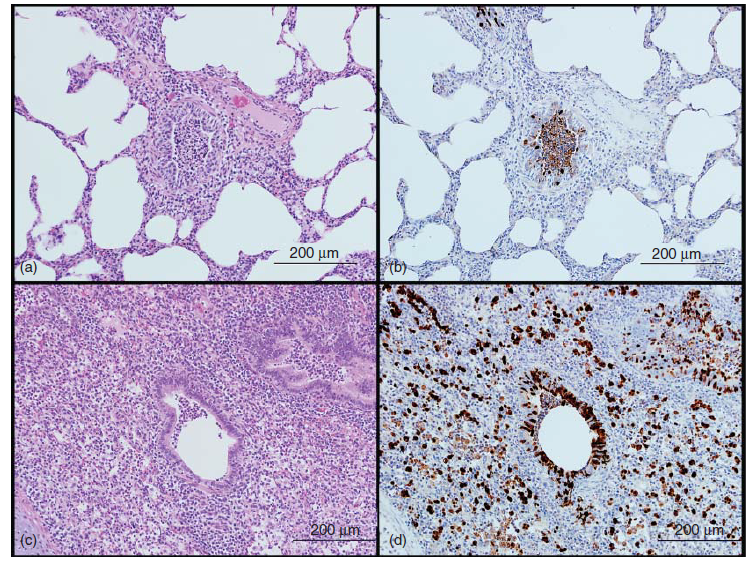
Gambar 16.2 Inokulasi eksperimental pada babi umur 4 minggu dengan virus H3N2 A/swine/Texas/4199-2/1998 48 jam setelah infeksi (PI). Sumber: Susan Detmer. GAMBAR 16.2A. Bronkiolitis berat nekropurulen dengan pneumonia intersittial; hematoksilin dan eosin (H&E), 200X. Gambar 16.2b. Sel epitel bronkioli moderat dan immunoreaktivitas intraluminal terhadap nukleoprotein anti-influenza A; imunohistokimia (IHC) dengan diaminobenzidin (DAB), 200X. Gambar 16.2c. Bronkiolitis nekropurulen dengan pneumonia alveolar berat (konsolidasi lobular); H&E, 200X. Gambar 16.2d. Immunoreaktifitas yang kuat pada alveoli dan bronkioli terhadap nukleoprotein anti-influenza A; IHC dengan DAB, 200X.
Patologi makros
Lesi makroskopik penanda bronkopneumonia cranioventral merefleksikan lesi mikroskopis dan rute infeksi. Rute aerosol dari infeksi berakibat pergerakan virus melalui sistem konduksi (saluran hidung dan trakea) ke carina, dan kemudian menyebar kedalam cabang-cabang pendek pohon bronkial dalam lobus kranial dan aksesorinya disebabkan tarikan gravitasi. Lesinya juga bisa memanjang sampai bagian paling kranial dari lobus kaudal paru. Selain itu, anomali anatomi bronkus trakea pada babi dapat menimbulkan lebih banyak lesi dalam lobus cranial pulmo dexter [13].
Meskipun beberapa lesi dapat terlihat dalam waktu 48 jam setelah PI, lesi makros belum berkembang secara penuh dan hanya dapat mewakili setengah dari lesi mikroskopis yang telah terjadi [13]. 5 hari setelah PI, pola lobular multifokal sampai lesi yang menyatu lebih konsisten dalam merefleksikan lesi mikroskopis. Sebagai contoh, keberadaan lobular ateletaksis terlihat secara makros sebagai wilayah poligonal konkaf, berwarna merah tua yang keras dan tebal ketika diraba (Gambar 16.3). Lobular dan lobar ateletaksis juga terlihat pada infeksi M. Hyopneumoniae, dan secara makroskopis bisa saja tidak dapat dibedakan dengan infeksi virus AI. Area poligonal yang terlihat membesar, berwarna merah tua sampai ungu yang sering dideskripsikan sebagai teraba seperti “hepar” atau memiliki tampilan seperti “daging” (meaty) merefleksikan konsolidasi alveolar (celah alveolar yang dipenuhi debris selular, sel radang, dan cairan edema). Lesi makroskopis ini lebih merupakan karakteristik infeksi bakteri aerosol dalam paru-paru dan dalam kasus infeksi AI sering dihubungkan dengan PRDC atau infeksi sekunder bakteri.
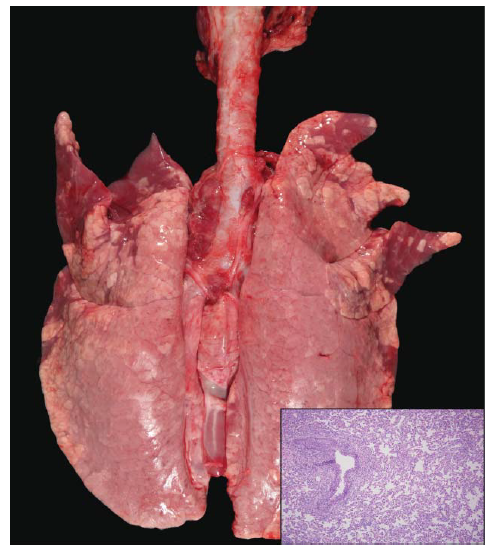
Gambar 16.3 Lesi makroskopis dalam paru-paru pada babi berumur 4 minggu yang secara eksperimental diinokulasi dengan virus H1N1 A/swine/illinois/02450/2008 5 hari setelah PI. Lesi lobular yang saling menyatu berwarna merah tua terletak di bagian cranioventral paru-paru, dan lesi mikroskopis (dalam insert) menunjukkan ateletaksis. Sumber: Susan Detmer.
Imunohistokimia
Imunohistokimia (IHC) mendeteksi antigen virus dalam jaringan. Dua antigen yang paling sering terdeteksi baik dalam jaringan beku maupun jaringan yang sudah difiksasi formalin adalah nukleoprotein (NP) dan hemagglutinin (HA). Antibodi melawan NP dapat digunakan untuk mendeteksi semua subtipe virus AI, akan tetapi antibodi melawan HA bersifat subtipe spesifik dan dapat menimbulkan reaksi silang dalam subtipe yang sama. Antigen NP terletak dalam nukleus dan sitoplasma sel yang terinfeksi [20, 23, 39, 84], sementara antigen HA terletak dalam sitoplasma dan di sepanjang permukaan sel [20]. Lokasi antien dalam jaringan akan tergantung pada lokasi replikasi virus. Jika infeksi utamanya terkumpul dalam bronkioli, imunoreaktifitas akan ada terutama di sel epitel bronkus dan dalam netrofil dan debris sel yang mengalami nekrosis dalam lumen bronkioli (Gambar 16.2). Jika infeksi menyebar dari bronkioli kedalam alveoli maka juga akan ada immunoreaktivitas, terutama dalam sel pneumosit tipe II yang bisa saja atau bisa terkelupas di sdalam celah alveolar (Gambar 16.2d).
Transmisi dan Epidemiologi virus influenza A pada babi
Transmisi
Pada babi, periode inkubasi setelah infeksi sampai pendedahan virus terjadi sangat singkat. Dalam infeksi eksperimental, 1 kali dosis inokulasi intratrakeal sebanyak 10 6-8 dosis infeksisu dalam kultur jaringan 50 (TCID50)/mL atau dosis infeksius untuk telur 50 (EID50)/mL biasanya akan berakibat pendedahan virus melalui hidung dengan titer sebanyak 10 2-4 TCID50/mL dalam waktu 24 – 48 jam PI [6,12, 69]. Hal ini sedikit bervariasi antar virus yang berbeda, tergantung pada kemampuan virus untuk menginfeksi sel, status kekebalan tubuh hewan, dan tingkat replikasi virus in-vivo.
Meskipun inokulasi intratrakeal lebih efisien dan konsisten dalam model eksperimental, dan mode transmisi yang sebenarnya dalam infeksi alami pada awalnya melalui saluran hidung, melanjut ke dalam sistem konduksi dan akhirnya ke paru-paru. Hal ini disimulasi secara eksperimental dengan menginfeksi secara kontak langsung dan melalui paparan ruang udara yang sama dengan babi terinfeksi yang sedang mendedahkan virus [1, 45, 69]. Lesi makros dan mikros yang diproduksi oleh transmisi virus pada model tidak bisa dibedakan dengan yang terlihat pada babi yang diinokulasi secara intranasal yang digunakan sebagai sumber infeksi [1, 69].
Epidemiologi
Dibawah kondisi lapangan, transmisi alami virus AI dapat terjadi sepanjang tahun, dan terdapat sejumlah faktor resiko yang memudahkan transmisi virus dan sirkulasi virus secara terus menerus satu atau lebih virus AI dalam satu peternakan atau dalam suatu sistem peternakan. Besarnya ukuran kawanan babi atau banyak jumlah babi per pen/sekat kandang adalah faktor resiko yang telah diketahui dengan jelas [15, 52, 65].
Salah satu dari faktor resiko paling penting yang berhubungan dengan tingkat deteksi virus AI yang tinggi adalah tipe peternakan, dan lebih spesifik lagi merujuk pada tipe peternakan farrow-to-finish (FTF) [10]. Pada peternakan FTF, babi finisher lebih mungkin ditemukan virus AI pada swab nasalnya jika mereka berada dalam peternakan yang sama dengan kawanan babi betina (sow herd). Dalam situasi ini, babi dari semua kelompok umur pada peternakan yang sama, meskipun telah dipisahkan dalam beberapa rumah kandang yang terpisah (bar), biasanya menggunakan manajemen aliran kontinyu (continuous -flow). Dalam peternakan aliran kontinyu, babi muda dikumpulkan bersama babi yang lebih tuan, menciptakan situasi dimana babi yang rentan terus menerus masuk ke dalam populasi dan mempertahankan keberadaan virus AI.
Pada beberapa peternakan FTF, babi-babi dipisahkan berdasarkan umur untuk menciptakan kelompok yang lebih homogen atau lebih stabil secara patogen. Homogenisasi ini terganggu saat babi-babi “fall-backs” (babi-babi yang ukurannya lebih kecil dari rata-rata ukuran babi dalam kelompoknya) dikembalikan lagi ke dalam satu atau dua kelompok untuk memberikan mereka waktu bertumbuh sebelum akhirnya dikirim ke penyembelihan. Wabah-wabah AI kecil dalam ruang-ruang dapat terlihat pada babi finisher ini. Dalam manajemen peternakan lainnya yang disebut all-in all-out (AIAO), seluruh babi dalam kandang ditempatkan pada waktu yang sama, diberikan waktu untuk bertumbuh kemudian dikeluarkan. Dalam salah satu studi, kurangnya pergerakan dalam sistem AIAO dihubungkan dengan meningkatnya resiko infeksi virus AI [15], dan studi yang lain menemukan bahwa manajemen AIAO berhubungan dengan resiko yang lebih rendah dibandingkan dengan manajemen aliran-kontinyu [10].
Kondisi lingkungan yang lebih memungkinkan terjadinya transmisi aerosol adalah temperatur dan kelembaban yang rendah [47]. Virus AI dapat dideteksi dalam udara di dalam dan diluar kandang (barn) baik pada hewan yang diinfeksi secara eksperimental [11] dan selama terjadinya wabah di lapangan [8]. Temperatur luar ruangan dan kecepatan angin yang ideal untuk terjadinya transmisi aerosol antar barn perlu untuk ditentukan, akan tetapi virus dapat terdeteksi sampai 2,1 km arah angin dari peternakan yang terinfeksi [10]. Temuan ini memberikan kredensial kemungkinan penyebaran secara aerosol antar peternakan. Saat ini hanya ada bukti anekdotal pada kasus-kasus dimana strain virus berpindah antar peternakan dan satu-satunya hubungan epidemiologinya hanyalah kedekatan lokasi [12].
Faktor-faktor risiko lain yang perlu diinvestigasi secara menyeluruh meliputi efektivitas vaksin babi untuk strain endemik, dan status vaksinasi dan kesehatan orang-orang yang bekerja di peternakan. Vaksin, baik komersial maupun autogenik (yang akan didiskusikan lebih detail pada Bab 19), digunakan secara luas pada babi. Kebanyakan vaksin itu digunakan pada babi betina (sow) agar muncul MDA untuk melindungi babi selama periode sapih dan periode awal lepas sapi (nursery). MDA biasanya menghilang antara umur 8 sampai 12 minggu – kisaran umur dimana lebih sering ditemukan virus dalam studi surveilans aktif [9]. Dengan kecocokan homolog yang pas, perlindungan lengkap melawan infeksi virus AI telah teramati [69], akan tetapi lebih sering dipergunakan vaksin heterolog yang memberikan perlindungan parsial [1, 12, 69]. Sangat penting untuk mempertimbangkan apakah vaksin dan penggunaannya secara tepat maupun tidak tepat, memainkan peranan memacu evolusi, dan apakah protokol vaksin yang saat ini digunakan telah menciptakan faktor resiko baru.
Selain faktor resiko, kekhawatiran epidemiologi lainnya adalah variasi regional yang terjadi pada virus AI, dan pergerakan virus yang mengikuti pergerakan babi [59]. Variasi regional karena antigenik shift dan drift lebih banyak terjadi dimana kepadatan babi rendah dan level pergerakan babi ke wilayah itu rendah. Salah satu faktor yang menentukan strain mana yang mendominasi dalam suatu wilayah adalah pergerakan babi. Dalam negara Amerika Serikat, pergerakan babi memegang peranan dalam keragaman virus AI yang ditemukan di daerah lubang serapan (sinkhole) ekologi wilayah Midwest yang merupakan destinasi akhir pemasaran babi dan wilayah dengan kepadatan populasi babi tertinggi [59]. Importasi virus AI yang terus menerus melalui importasi babi dari wilayah lain ke wilayah Midwest telah berakibat munculnya varian yang sangat berbeda secara genetik saling bersamaan bersirkulasi dan bertukar segmen gen melalui reasortansi [59]. Varian ini meliputi tujuh garis keturunan yang memiliki hemaglutinin berbeda yaitu, H1 alfa, beta, gamma, lamda1, lamda2, pdm09, dan klaster 1V H3 [2, 44]. Terdapat beberapa kelompok antigenik dalam klaster IV H3 yang tidak mengikuti pola pengelompokan subgrup secara filogenetik [42]. Analisa lebih jauh diperlukan untuk menentukan apakan terdapat prevalensi regional untuk klaster antigenik baru ini.
Transmisi Zoonosis
Zoonosis memainkan peran kunci dalam epidemiologi dan evolusi virus AI pada babi. Transmisi virus AI musiman dari manusia ke babi telah terdokumentasi dengan baik [58, 60]. Untuk alasan ini, strategi pengendalian untuk memitigasi risiko yang dimiliki manusia terhadap kawanan babi harus dipertimbangkan. Meskipun permasalahan yang sama tentang vaksin heterolog yang menghasilkan perlindungan parsial dapat mengurangi gejala klinis dan pendedahan virus, disisi lain vaksin musiman juga harus dipertimbangkan selain juga penggunaan respirator N95 untuk mengurangi resiko zoonosis dari manusia ke babi dan babi ke manusia. Virus asal unggas kadang-kadang masuk ke dalam populasi babi yang dikandangkan yang berasal dari unggas domestik maupun liar, akan tetapi peternakan multispesies dan babi yang dipelihara di luar lebih beresiko [16].
Meskipun kebanyakan zoonosis yang telah terdokumentasi melibatkan transmisi virus H1N1pdm09 dari manusia ke babi, yang menjadi menetap sebagai virus musiman pada manusia setelah pertama kali muncul sebagai pandemi di tahun 2009, strain virus AI musiman pada manusia ditemukan secara sporadis ditemukan bersirkulasi pada babi [60]. Infeksi dari babi ke manusia juga telah terdokumentasikan secara sporadis. Terlepas dari H1N1pdm09, infeksi virus AI asal babi pada manusia biasanya adalah akibat dari transmisi terbatas dari manusia ke manusia, dan orang-orang yang terinfeksi biasanya memiliki riwayat terkini terpapar babi [7, 19, 38]. Satu pengecualian adalah virus varian H3N2 yang dihubungkan dengan paparan langsung atau tidak langsung pada saat penyelenggaraan festival agrikultur di Amerika Serikat [14].
Resiko terbesar yang dimiliki oleh infeksi zoonosis ini adalah kemungkinan babi menjadi penyedia kesempatan reasortansi antara strain virus AI manusia dan babi. Virus pandemi yang berasal dari babi yang muncul di tahun 2009 menggarisbawahi pentingnya pemahaman tentang patogenesis, transmisi dan evolusi virus AI baik pada manusia dan babi.
DISCLAIMER
Artikel terjemahan ini dimaksudkan bagi pembaca berbahasa Indonesia untuk dapat mempelajari artikel keilmuan terkait. Penerjemah tidak mendapatkan keuntungan apapun dari kegiatan penerjemahan ini.